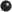 In table 2, one can find the
optimized geometries (Degree and Angstrom),
UHF energies (Hartree), ionization potentials (IP in eV),
and dipole moments (DIP in Debye)
of H3O.,
obtained with various basis sets.
In table 2, one can find the
optimized geometries (Degree and Angstrom),
UHF energies (Hartree), ionization potentials (IP in eV),
and dipole moments (DIP in Debye)
of H3O.,
obtained with various basis sets.
Spin contamination was found to be small, for example
< S� >
=0.752 for D95**/R3 ,DH/R8 and VD/R7.
Table 2. Optimized geometries
| Basis set |
Species |
Symmetry |
H-O-H (degree) |
O-H ( �) |
Energy (A.U) |
IP (eV) |
Dip. moment (Debye) |
| 4-31G/R1 |
H3O. |
C3v |
118.9 |
0.995 |
-76.375 |
4.84 |
0.249 |
| 4-31G/R2 |
H3O. |
D3h |
120 |
0.997 |
-76.378 |
4.86 |
0.009 |
| D95**++ |
H3O. |
C3v |
108.6 |
0.986 |
-76.504 |
4.89 |
0.471 |
| D95**/R3 |
H3O. |
C3v |
108.67 |
0.986 |
-76.505 |
4.89 |
0.806 |
| D95**/R4 |
H3O. |
C3v |
108.64 |
0.987 |
-76.506 |
4.915 |
0.7947 |
| D95**/R4 |
H3O+ |
C3v |
113.66 |
0.963 |
-76.331 |
|
|
| VD/R5 |
H3O. |
C3v |
108.19 |
0.983 |
-76.519,071 |
4.89 |
0.837 |
| VD/R6 |
H3O. |
C3v |
108.18 |
0.982 |
-76.519,326 |
4.99 |
0.857 |
| VD/R7 |
H3O. |
C3v |
108.56 |
0.982 |
-76.521,061 |
4.88 |
0.847 |
| DH/R8 |
H3O. |
C3v |
108.58 |
0.984 |
-76.506,763 |
4.89 |
0.783 |
| DH/R8 |
H3O. |
D3h |
120.00 |
0.981 |
-76.501,290 |
4.77 |
0.0 |
From these tables, it is possible to acertain the influence
of the basis set quality.
Polarization functions are absolutely necessary for
obtaining a pyramidal geometry.
When considering pyramidal geometries, with almost the same
angles, the dipole moment varies from 0.4709 Debye to
0.857 Debye. Extra diffuse functions have a dramatic
effect on the dipole moment value for example
considering D95++** and D95**/R3, while the total
energy varies little.
The ionization potentials (IP) were obtained via the Koopmams' theorem,
and correspond to a good
approximation to the vertical detachment energy (VDE) of
the outer electron.
The adiabatic detachment energy (ADE) was also computed, taking
into account the fully relaxed H3O+ cation geometry and is
lower than the VDE.
For the D95**/R4 basis set, since we optimized
also the parent hydronium cation, it is possible to compare the
electron affinity (EA = 4.637 eV) of the cation,
the vertical detachment energy (VDE=IP= 4.89 eV) and the
adiabatic detachment energy (ADE= 4.76 eV)i of the hydronium radical.
Gellene and Porter
[Gellene 84]
estimated that the electron
affinity (EA) of H3O+ should be equal to the IP
of H3O..
We can see that there is however a difference of 0.278 eV between the
computed IP and the computed EA. One part of the difference may due
to geometric changes, an another part may be related
to electronic relaxation which
is not taken into account within the framework of Koopmans' theorem.
The IP is not
necessarily higher, with a better basis set ( VD/R6 versus VD/R7).
The more economical DH/R8 basis set features an IP value very
similar to the IP value 4.88 eV of our most expansive basis set.
The inversion barrier, corresponding to the umbrella motion,
was estimated around 3.43 Kcal/mole by optimizing the H3O. geometry
under a D3h symmetry constraint. It appears therefore that
this molecule is more floppy than ammonia (inversion barrier 5.8 Kcal/mole).
In relation to a Fermi contact point ESR analysis,
we list the atomic spin densities in table 3.
The spin densities on hydrogen atoms belonging
to the H3O.
radical are small but positive.
It is interesting to notice that the spin densities on the
hydrogen atoms seems to be quite sensitive to the geometry.
In D3h
planar or nearly planar geometries,
the spin density value nearly
triples. We report also the spin densities at the
dehydrogenation
transition state geometry, on the H2O and H.
fragments.
Table 3. Atomic spin densities (Atomic units)
| Basis set | Geometry | Oxygen | Hydrogen |
| 4-31G/R1 | C3v |
0.768 | 0.016 |
| 4-31G/R2 | D3h |
0.798 | 0.018 |
| D95++** | C3v |
0.847 | 0.005 |
| D95++**/R4 | C3v |
0.836 | 0.005 |
| VD/R6 | C3v |
0.806 | 0.005 |
| VD/R7 | C3v |
0.809 | 0.005 |
| DH/R8 | C3v |
0.760 | 0.005 |
| | D3h |
0.926 | 0.012 |
| |
TST H2O |
0.807 |
0.001 |
| |
TST H. |
|
0.071 |
It is crucial
to examin the MO graphic of the outer electron in order to
grasp its physical nature. In our specific case
the symmetry-adapted (4A1) HF MO
(fig. 1) or the
corresponding natural orbital (NO)
(fig. 2) are similar,so
we may consider the graphical plot of one or the other.
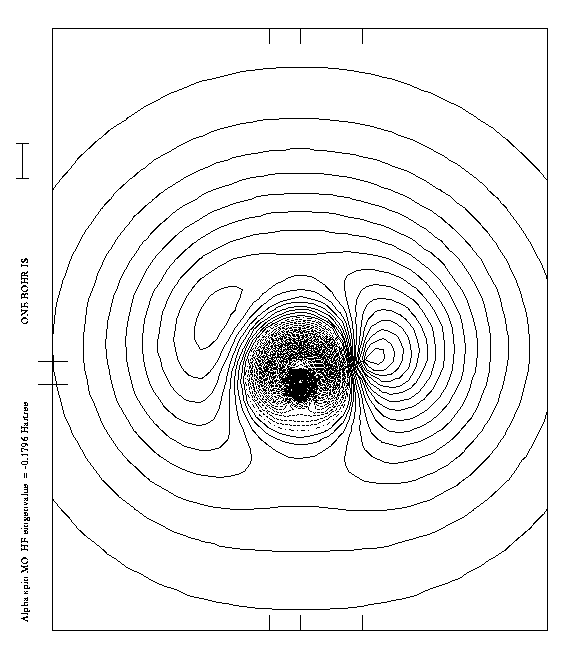
Figure 1.
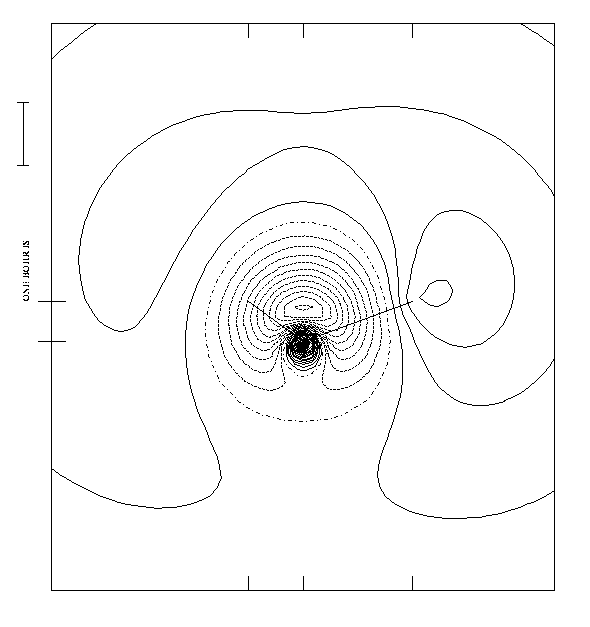
Figure 2.
 This NO wavefunction (cf fig 2) features two clearly distinguishable
nodal surfaces ( - . - . -) which separate out regions of
alternative phase sign. So it is possible to
assimilate grossly this NO and HF MO to a 3s-type supermolecular
orbital.
However the supermolecular perspective, though well
adapted to spectroscopy, does not give any insight if
the outer MO is a non-bonding MO or an antibonding MO.
This NO wavefunction (cf fig 2) features two clearly distinguishable
nodal surfaces ( - . - . -) which separate out regions of
alternative phase sign. So it is possible to
assimilate grossly this NO and HF MO to a 3s-type supermolecular
orbital.
However the supermolecular perspective, though well
adapted to spectroscopy, does not give any insight if
the outer MO is a non-bonding MO or an antibonding MO.
 We can see that the outer nodal surface goes between the
oxygen atom and the hydrogen atom , therefore the outer MO is
an antibonding orbital and as such cannot be
assimilated to a pure Rydberg orbital .
On the other hand, the outer orbital is somewhat special,
by comparison to other valence bonding or antibonding
orbitals, because it is so diffuse.
We can see that the outer nodal surface goes between the
oxygen atom and the hydrogen atom , therefore the outer MO is
an antibonding orbital and as such cannot be
assimilated to a pure Rydberg orbital .
On the other hand, the outer orbital is somewhat special,
by comparison to other valence bonding or antibonding
orbitals, because it is so diffuse.
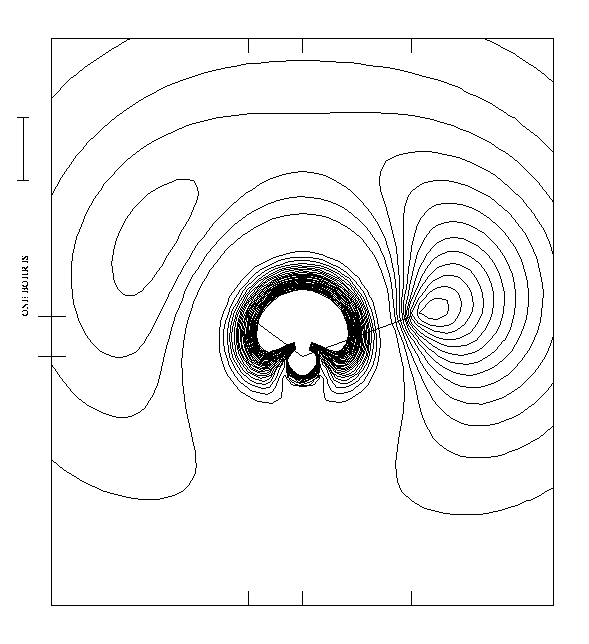
Figure 3.
On the total electron density map (cf. fig 3 ), we can
clearly distinguish a high electron density region pertaining to
the
H3O+ core, and
a low density, diffuse region related to the unpaired outer
electron.
One way to assess this diffuse character is to compare
with orbitals of isolated atoms. The
H3O. radical
is isoelectronic to sodium (Na), which comprises
a lone electron in a 3 s orbital (cf fig 4).
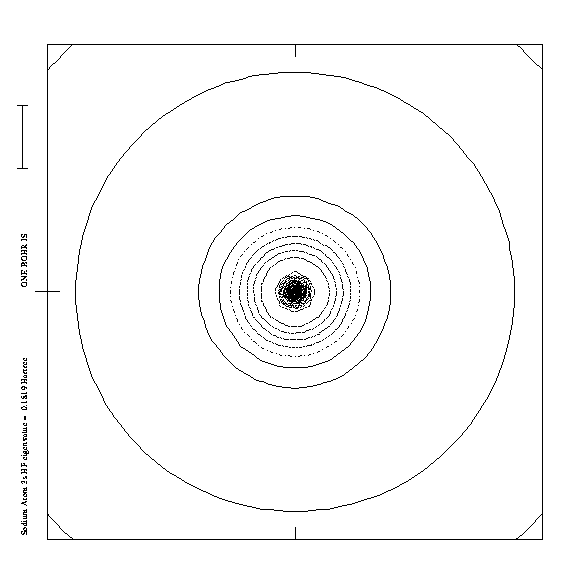
Figure 4.
Remarkably enough the 3s electron of the sodium atom is
the most diffuse outer electron of all atoms.
One can easily check that the various
Na basis sets feature the smallest
gaussian exponents, by consulting a handbook
[Poirier 85]
of gaussian basis sets.
For convenience, we are going to designate, when we want to
stress the diffuse character,
the outer MO as
the low-lying Rydgerg orbital, or the outer electron as
the Rydberg electron, although we must keep in mind the
antibonding character.
An antibonding character does not contribute to molecular
binding, but it is yet another question to
determine if this antibonding character is going to strongly
undermine the electronic glue built by inner bonding
MOs.
We suggest
that an easy and intuitive way to judge of the dissociative
or the non-dissociative character of an antibonding
molecular orbital, is to inspect
MO graphics with a sufficient contour resolution.
A totally nondissociative antibonding MO occurs when the nucleus
position coincides with an electronic distribution barycenter.
However, in the present case, we can measure on the graphics plot,
that the maximum of the electronic density created in the third
phase region of the NO or the 4A1
wavefunction, is situated approximately
0.14 � to the exterior of the hydrogen atom or 1.12 � from
the central oxygen atom.
(cf fig. 2, fig 3).
In order to lower its energy within the specific outer wavefunction,
the Hydrogen atom will tend to be propelled outside, towards
this electron density maximum.
As we shall see later,
it is quite interesting to see that precisely this distance of 1.12 �
corresponds to the O--H. distance in the transition state
geometry towards dissociation.
Considering now an isolated hydronium radical
in condensed phase, we might expect that,
because of electronic repulsion
due to surrounding electrons from neighbour molecules,
the barycenter in the third phase region
in the 4A1 MO might be displaced towards
the proton, therefore reducing the dissociative character of the
4A1 orbital, and would
contribute to increase the dissociation barrier
of this radical in condensed phase.



